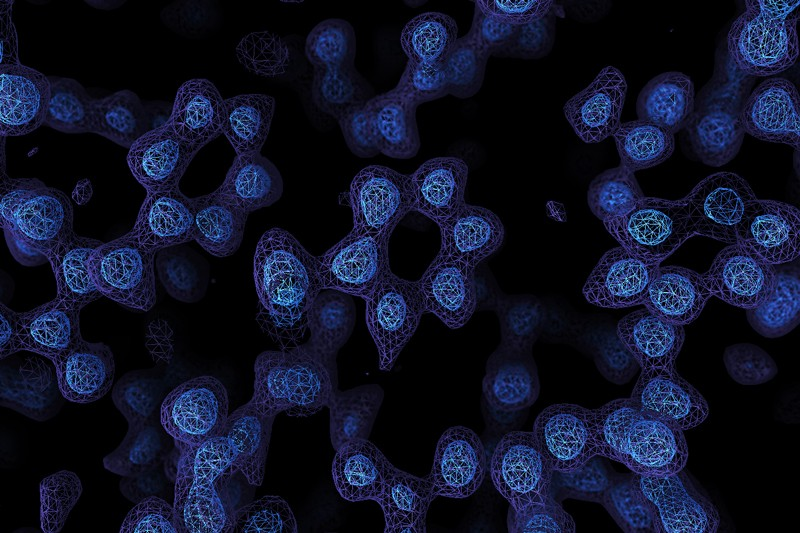
There’s an old joke that you can’t trust atoms — they make up everything. But until fairly recently, there was no real way to see individual atoms. You could infer things about them using X-ray crystallography or measure their pull on tiny probes using atomic force microscopes, but not take a direct image. Until now. Two laboratories recently used cryo-electron microscopy to directly image atoms in a protein molecule with a resolution of about 1.2 x 10-7 millimeters or 1.2 ångströms. The previous record was 1.54 ångströms.
Recent improvements in electron beam technology helped, as did a device that ensures electrons that strike the sample travel at nearly the same speeds. The latter technique resulted in images so clear, researchers could identify individual hydrogen atoms in the apoferritin molecule and the water surrounding it.
For years, the standard way to study protein structure was to form a crystal and study the way that crystal diffracts X-rays. However, some proteins are difficult or even impossible to crystalize. Cryo-electron microscopy doesn’t have this issue. The microscope operator has to flash freeze the sample. A better understanding of protein structure can further research into things such as enzyme action and help scientists develop better drugs.
Computer analysis of the electron emissions is a key part of the technique as well and one of the scientists involved believes that resolutions below 1 ångström are probably not possible for this method with current computing power. In addition, the quality of the image depends partially on the stability of the protein. Apoferritin is highly stable, but some other molecules they tested are not that stable. That means X-ray crystallography will probably remain the method of choice for proteins that will easily crystalize. This is especially true since the cryo-electron microscopy method can take hours or days of data collection to form a complete image.
If you want to know more about how an electron microscope works, we’ve talked about that before. If you want to build your own atom-resolving microscope, check out our survey of builds.













Read it again Al, a map is not a direct image. They infer the shape from computation.
So, kind of like radio astronomy. Or crystallography. Or computed tomography. Or magnetic resonance imaging. Or positron emission tomography. Or single photon emission computed tomography. Or ultrasound imaging or seismography. Or synthetic aperture radar. And lots more ways to make “images”. Should we call them all “maps” too?
I concur that those are images, but none are direct images.
How exactly is a radio astronomy “image” somehow less “direct” than a CCD image? Or are those not “direct” either? Is this some weird photographer’s equivalent of vinyl vs digital? “If it ain’t formed directly on the thing, it ain’t a direct image!”
Radio astronomy images are constructed by recording amplitude and phase and mapping that to intensity at different angles. CCDs record amplitude at different positions which are scanned over, corrected, and mapped to intensity from different angles. Both require math to map them to an image. In one case the mapping’s a little easier, but it’s still math.
I would say that direct imaging is where all the information required to define a data point is gathered at that data point.
So, if I choose to make an image using an electronic detector and a physical refractive or reflective element to do the transform prior to detection, it counts as a “direct” image, but if I choose to do the transform computationally after detection it does not?
I could place an array of radio receiver antennas at the focus of a large dish reflector (just like an optical imager with a mirror lens), and produce a “direct” image, or I could omit the focusing dish and do the beamforming electrically by varying lengths of cable, or I could do the beamforming computationally. Which one counts as an “image” or “direct image”?
It is convenient that we have inexpensive and simple real-time optical transform processors (“lenses”) for visible photons. For other energies (or wave-like particles like electrons, as appropriate), we have to turn to more complex techniques, but it’s all a matter of degree, isn’t it? At what point does an “image” cease to be a “direct image”?
Even a pinhole camera can be considered an optical processor. Clearly, if we use a pinhole to produce pictures with 2 eV photons (green light) it would count as an “image”. If we use the same pinhole for 140 keV photons in a SPECT camera does it not count as an “image” just because a computer is in the path?
Welcome to real life, there’s a million edge cases.
It’s a big universe. There is no edge.
“I would say that direct imaging is where all the information required to define a data point is gathered at that data point.”
That’s not how optics works. The very existence of an aperture means a single point turns into an Airy disk. In other words, the information required to define the intensity at a single point is gathered at *multiple* points.
You’re making an arbitrary distinction.
The claim that “there was no real way to see individual atoms” is patently false. While true for atoms in large protein molecules, SFM (scanning force microscopy) has been able to resolve individual atoms on a surface for a couple of decades now. Saying SFM doesn’t count as “seeing” individual atoms is absurd. The described method is likewise achieved computation, not by “seeing” the atoms. Pretty disappointing reporting from a site that should know better.
I don’t blame HAD, the parent article’s similarly vague. The key isn’t that *atoms* have been “imaged” for the first time, it’s that *bio* molecules have been – fragile ones that don’t crystallize or lend themselves to STM/AFM surface prep. Obviously STMs have resolution hundreds of times better than this, but you cant use a ln STM to study proteins
if this is possible why is ‘foldig@home’ still a thing? why not just image the proteins of interest?
Folding@Home does not image proteins. It tries to fold them into new and useful configurations.
This is done at cryo temps, when the atoms are “frozen.” There’s literally no way to image how a protein *interacts*
I would presume though that if you flash freeze a sample at the half life of a reaction time, you would get a cross section of reacted, not-reacted and various stages in the process of reacting, which would allow inference.
Possibly, although having a somewhat amorphous sample might complicate the actual measurement itself. I’m not sure about cryoelectronmicroscopy, but for things like x-ray crystallography you need a fairly uniform set of molecules in a crystalline lattice.
This is at a quantum level: you don’t have “various stages in the process of reacting.” The molecules flash-freeze into the available free-energy minima. You won’t get transient states, because, well… they’re not stable. Worse, some of the observed states might not even occur in a normal reaction because the transition rate from the transient states to the *real* final state might be high enough that at typical temperatures, those “intermediate” states never really get populated. But due to the flash freezing, the transient states fall there and can’t get out.
Basically, imagine two mountains with a shallow valley in the middle and deep valleys on the ends. Now imagine that you’re on the Moon: you can easily leap up to the first mountain, over to the second, and down to the other end, and never spend time in that “middle” valley at all. But imagine that as you’re going from one end to the other, suddenly gravity increases 100-fold. You’ll never end up at the top of the mountain, but you *will* end up in that “middle valley” that you never actually ended up in the first place.
Cryo-EM will get you the structures of the stable states. It won’t give you *any* information about the path between them, because you’ve literally *killed* those states to freeze the molecule so you can study it.
This is a fundamentally unbeatable problem – you can’t observe complex molecules interacting without disturbing the overall process somehow. Maybe you’ll get lucky and you can alter the situation only a little, but in the end you’re still disturbing it.
Good description, and good way to picture it. Kind of a like a simulated annealing problem.
Yes, but I was under the impression that between a native protein state and that which it has folded completely to react, that there were multiple semi stable intermediate states…
https://www.pnas.org/content/111/45/15873.figures-only
“Yes, but I was under the impression that between a native protein state and that which it has folded completely to react, that there were multiple semi stable intermediate states…”
They’re a *transition* state, which is why they’re labelled “TS” in that figure – they’re at a higher energy potential than the states around them, which means once you flash-freeze it there’s no way they can end up there. They’re not ‘semi-stable’, they’re *unstable*. And part of the problem is that at normal temperatures, certain states (the little valleys visible on that diagram) *are* actually semistable, but when you flash-freeze them they’re *stable*. So you end up with a state that might not even exist in the reaction pathway and thinking somehow it is involved.
Good summary here:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3785246/
“In a nutshell, what has been said in the section above implies both potential and limitations of cryo-EM. Its potential lies in the fact that multiple states, corresponding to the local energy minima, may be simultaneously recovered from the same sample – in other words, the structural inventory introduced before. The limitations relate to the transitions among the “basin” states: it must be emphasized that information about these transitions is entirely out of reach for cryo-EM.”
…
“However, the fact that the molecule in states that correspond to places with high gradient in the energy landscape cannot be captured by 3D visualization remains unchanged.”
…
“The missing information about pathways connecting the different observed states evidently has to come from elsewhere, namely: (i) results of other experimental techniques, (ii) topological considerations, or (iii) computational simulations.”
The ‘other experimental techniques’ there involve stuff like attaching portions that flouresce to the protein and monitoring how those portions move, so again, basic fundamental limitation – you can’t observe without altering. Hence the importance of accurate computational simulations.
These days, for the most part, obtaining an accurate 3D structure of the (lowest energy) structure of a protein is fairly routine if you have a crystalline sample and a decent x-ray diffractometer (although it needs a bit of skill, etc. ). It is extremely difficult, however, to try to obtain native proteins in structures other than the lowest energy one (unless you bind other molecules to them, which obviously changes everything). So, the folding@home project tries to use computational methods to find other structures that are still reasonably low energy enough to be feasible, which might provide alternative binding sites for drugs etc.
In more often used units 1.2 x 10-7 millimeters = 0.12 nanometers = 120 picometers.
The angstrom ( ångström, Å , 10E−10 m, 0.1 nm ) is useful in this context, being about the radii of atoms. Deprecated unit, yes, but some people still use inches too.
The phrase “protein atoms” makes me wonder about the reliability of the article. Precision is not just something a mohel does.
It makes sense here though as the actual atoms forming the molecule is what can be seen. We’re below molecule scale.
Never heard about Raymond Rife microscope? It worked better than this, until government made it disappear.
https://sustainable-nano.com/2017/08/18/royal-rifes-universal-microscope-and-why-it-cant-exist/