Genetic defects are exceedingly common, which is not surprising considering just how many cells make up our bodies, including our reproductive cells. While most of these defects have no or only minor effects, some range from serious to fatal. One of these defects is in the CPS1 gene, with those affected facing a shortened lifespan along with intensive treatments and a liver transplant as the only real solution. This may now be changing, after the first successful genetic treatment of an infant with CPS1 deficiency.
Carbamoyl phosphate synthetase I (CPS1) is an enzyme that is crucial for breaking down the ammonia that is formed when proteins are broken down. If the body doesn’t produce enough of this enzyme in the liver, ammonia will accumulate in the blood, eventually reaching levels where it will affect primarily the nervous system. As an autosomal recessive metabolic disorder it requires both parents to be carriers, with the severity depending on the exact mutation.
In the case of the affected infant, KJ Muldoon, the CPS1 deficiency was severe with only a low-protein diet and ammonia-lowering (nitrogen scavenging) medication keeping the child alive while a search for a donor liver had begun. It is in this context that in a few months time a CRISPR-Cas9 therapy was developed that so far appears to fixing the faulty genes in the liver cells.
CPS1 Gene Deficiency
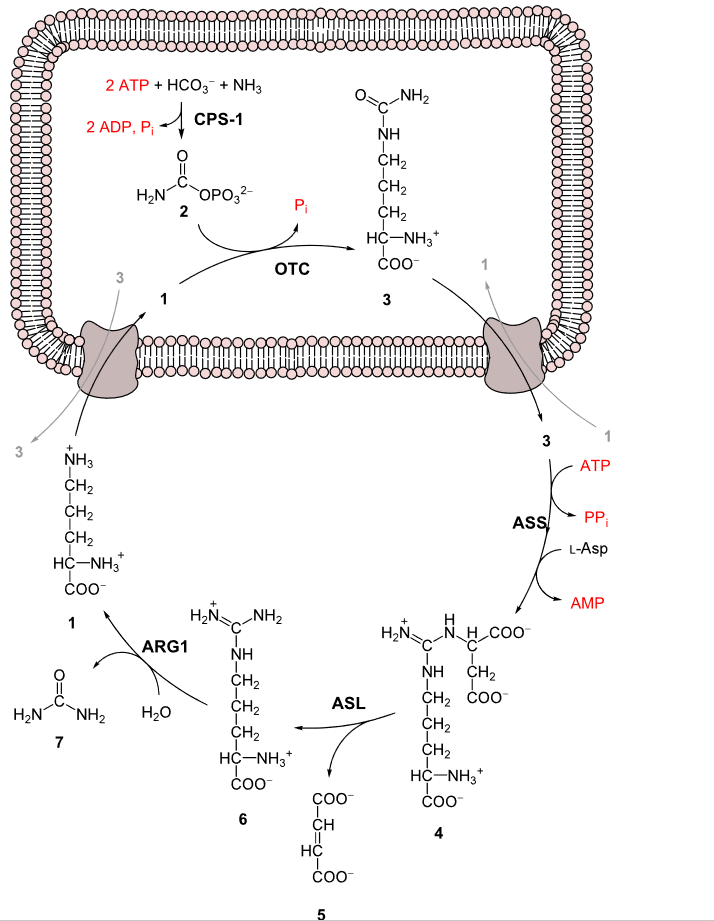
Despite its toxicity to living beings, ammonia (NH3) is an essential part of these same living beings, primarily in the form of amines (R-NH2), itself a rather indispensable part of amino acids, specifically the 22 proteinogenic amino acids from which proteins are formed. Just as ammonia is required for the amination process, so too is ammonia formed inside the body mostly as the result of transamination and deamination of these biogenic amines. This is a process that takes place primarily in the liver and involves the deamination of both the body’s own waste proteins as well as those from one’s diet.
Since only part of the ammonia can be reused for new amino acids, the rest has to be neutralized. Due to the toxicity of ammonia, blood levels have to be limited to <50 µmol/L or hyperammonemia will occur. This is where the urea cycle comes into play to maintain a healthy ammonia level.
The very first step of the urea cycle is the conversion of ammonia to carbamoyl phosphate:
NH3 + HCO−3 + 2ATP → 2ADP + Carbamoyl phosphate + Pi
Normally this is a very slow reaction, which is where the enzyme CSP1 comes into play as catalyst. In humans the gene for this enzyme is located on chromosome 2’s long arm, at locus 2q34. If there is a mutation in this gene that prevents it from working as a catalyst, ammonia levels in blood plasma will keep rising, eventually reaching levels where the nervous system is affected. In infants this is noticeable as lethargy, seizures and a lack of normal developmental milestones. Without treatment, developmental delay, intellectual disability or death affect 50% of babies.
Undoing A Mutation
When KJ was born on August 2024, it was noticed that he was lethargic, with stiff muscles and other worrisome symptoms. After a severe CPS1 deficiency was diagnosed via genome sequencing, KJ was hospitalized at only five months old. KJ’s only hope appeared to be a liver transplant and was put on the list for a donor organ, providing a slim hope at best. Meanwhile, a team of researchers started researching the cause of KJ’s CPS1 deficiency and the mutations behind it.
As described by Dr. Eric Topol in his summary of the (paywalled) paper by Gropman et al. in NEJM, both the father and mother were found to be carriers for CPS1 mutations, with the father carrying the truncating Q335X variant and the mother another (E714X). If either mutation could be corrected, the child would have one functional copy and theoretically be able to produce enough CPS1 to have a functional urea cycle without external assistance.
A complicating issue here is that despite the many reports of gene-editing with CRISPR the past years, there are various gradations, with what Dr. Topol refers to as CRISPR 1.0 through 3.0:
- CRISPR 1.0: A CRISPR-Cas9 tool causes sufficient double-strand damage to disable the gene (knock-out). Crude and not relevant here. Also performed ex vivo.
- CRISPR 2.0: Introduced single-strand cuts that allow for limited base editing, e.g. swapping A for a G.
- CRISPR 3.0. Expands base editing to include multiple base pairs, both ex vivo and in vivo.
These methods have previously already been used ex vivo to create modified T-cells for CAR T-cell immunotherapy in the context of cancer treatments. In terms of in vivo treatments, there is the 2023 knocking out of PCSK9 liver protein to reduce bad cholesterol levels and the more recent base editing of the PiZ mutation responsible for liver and lung damage. There’s also ARCUS, which is a viral vector-based method of base editing that has seen use in fixing another urea cycle-related disorder.
Although only CRISPR 2.0 was needed here, what was unique in the case of KJ was that this would be the first fully personalized base editing therapy, applied in vivo and developed within the span of a mere six months.
Crossing All The Ts

With how experimental this gene therapy for KJ’s CPS1 disorder was, the researchers had to go through the entire gamut of tests, including on animal models. With a base editor developed to target the father’s Q335X mutation and rewrite it to the correct base pairs, mice were bred that had the same CSP1 mutation, in addition to testing on non-human primates, all to validate the approach and gain FDA approval.
The base editor’s goal was to rewrite the the wrong bases at the Q335X location on locus 2q34. A concern with any application of CRISPR is so-called off-target edits, but the safety review seems to have passed here without serious issues.
Starting with a very low dose, blood plasma ammonia levels were carefully monitored with no noticeable changes. Three weeks later the second, higher dose was injected, with reportedly positive effects on the ammonia levels. A third dose was injected a while later, though the results of this aren’t know yet. In the absence of a liver biopsy it is hard to say in how far this is a true cure, as reported so far is a reduced need for medications.
Per reports, KJ is however doing better, hitting developmental targets and got over two viral infections, without an ammonia crisis. Further injections of the treatment will likely administered with an mRNA approach rather than the (presumed) virus vector used so far due to immunity concerns with a virus vector. Open questions remain regarding how many cells have been truly edited in KJ’s liver and what the overall effectiveness is.
This leads us to cautiously welcome this news as a step forward in personalized gene-therapy, while realizing that the road ahead for both KJ and the rest of us is still full of unknowns and challenges. That said, one can only hope for KJ’s best possible progress and ideally serving as a beacon of hope for others afflicted by genetic disorders like CPS1 deficiency.
Featured image: “CRISPR Cas9” by Ernesto del Aguila III, NHGRI, Courtesy: National Human Genome Research Institute














Amazing thing is the whole organism works as well and long as it does with as few problems.
With the complexity of the human organism the amazing thing is not how many people are sick but how few.
That’s what millions of years of evolution will do for you.
And now that we intervene so nature isn’t killing our defectives as efficiently as it used to, we look upon technological solutions to restore broken ones to working order.
billions of years of evolution*
If we can use technology to allow our “defectives” to live full and happy lives they are not “defective”. Our brains are a part of our evolution too.
Wait till you get older. Also, before science, we were considered old if we got to 40. A few did live till older but nothing like we do now. Again, science did this. Life is just one big hack-a-day experiment. :D
No thats a myth from misunderstanding statistics. Infant mortality was very high this cut average lifespans so low as 40ish but thats average for whole population where a lot of children didn’t survived infancy and toddler time to the point where some cultures waited with naming children till they are past that most dangerous age. Once you where past infancy and childhood into your teens you had good chance to live to about 60-70. Another thing cutting lifespans of women was childbirth once you where after your child bearing years it then good chance hitting up till low 70ties. 40 wasn’t considered old especially for men, 50+ was.
Right. Getting old was the same back then. Getting TO old age was the hard part. Lots more things could kill you back then.
Thank you. We need to keep pointing this out until the myth goes away :-)
The redditoid conception of “science” held by people like you is actually harmful to science.
I’m amazed they were able to do all the animal trials in this short time span.
I’d bet they already had as much in place as possible, the ethics committee were on alert, and they were just waiting for an eligible patient. Either that or they’re the most efficient research department in history.
Interesting times, this could be a really awesome technology, and also a really awful one.
That kind of makes me worried about the at-scale viability of this personal gene-editing. Since every patient will be the first and only recipient of a newly-developed drug, regulators would presumably insist on a full round of animal testing – as well they should, considering the repercussions of a gene-edit gone bad.
Does that mean we’ll have to set up mass-manufacture of bespoke model mice, one batch per patient? Entire zoos of primates dedicated to toxicity tests? My imagination veers into absurdity here, but I don’t see another way to safely handle these treatments at scale.
Unfortunately I don’t think you’re being absurd. I think that’s pretty likely, considering how many animals are killed for meat consumption, the only thing stopping society from doing this is the technological hurdles. :(
This isn’t the first human trial of CRISPR based gene editing, it’s building on a body of work.
That’s because–as usual–they didn’t
No, they did, this isn’t actually “new” per-say and the animal testing has been going on for years.
Even if this treatment is successful in the long term, the person’s germ cells may still contain the defective gene, which could be passed on to any progeny.
If genetic modification methods which are developed to fix this and other fatal genetic defects do not also fix the fatal genetic defect in germ cells, the fatal defects will then be passed on to successive generations.
This would likely lead to more and more individuals requiring genetic defect treatments in order to survive to adulthood.
Their germ cells will definitely still contain the defective genes. Germline editing is banned in the US, where the patient is from and was treated, as well as in many other countries.
Her treatment was not systemic, it was targeted, They used lipid nanoparticles to transport the viral vector to the liver where it was released when the liver broke down the lipid shell.
In this case the condition was an issue of a maternal genetic mutation AND a paternal genetic mutation, the absence of either would not have resulted in the condition manifesting. SO unless this childs grows up and marries someone with either her mother or her fathers mutation, or some other comparable mutation her offspring would not be at risk of anything more than carrying the potential risk along.
Change all she and hers to he and hims. My wife died in 2019 of the same cancer that Crispr cured in Emma Dimery in 2023 so in reading about Crispr research again, projection caused me to misgender this patient. My bad
I think the other issue is not knowing that you’re a carrier of the defective gene until after the child is born. Once you know you have it, it is possible and legal in some countries under some conditions to pre-screen embryos to select out that trait. At a quick look, the UK Human Fertilisation and Embryology Authority allows screening for hyperammonemia from a different genetic defect, so it doesn’t seem unreasonable that they might permit it for this one, too. CRISPR is more for people conceived before you know there is a genetic risk.
Screening is not modification. That screening is defined to check the viability of a fetish when there is reason to suspect a problem and it’s also done in the US. The major difference is that in the US the parents opt to bring a nearly brain dead child to term, then sue the hospital (even though they were warned) then get a large settlement. This is frequent enough someone I know left the field out of frustration after providing expert testimony in many cases.
And if genetic modification treatments continue to correct such recessive genes without altering germ cells, the probability of a couple both having the same recessive gene causing a fatal defect can only increase.
In the future, should couples have germ cell DNA tests done to see if they have identical recessive genes? Should it be while they are dating before they get serious? Before they are married? Before they decide on having children?
Or, on finding their unborn child has a fatal genetic defect, do they just butcher the kid in the womb, or spend the time and expense to have the child born and see if genetic modification will correct the problem (while leaving the same problem they faced for their child when grown up)?
This kind of screening is already common and sadly people frequently elect to have the child anyway, even if they have been informed they will be crippled. That’s what you get when people value birth over life.
It’s probably preferable to be alive and abstain from having biological children, compared to dying. Especially for men, having your child conceived through the use of donated sperm shouldn’t really be that huge of an issue.
As a man, I’m sorry, but I want my children to carry both my genetic code, and the one of my wife.
I don’t wee why it “shouldn’t really be that huge of an issue” for the men to stop passing their genetic code.
If you really want to edit code, just do in vitro fecundation and correct only the lethal mutation in the sperm, that way, (almost all) your genetic code is passed, and your child is healthy.
Eugenics shouldn’t come with complete loss of your code.
It shouldn’t but unfortunately real life doesn’t conform to your moral expectations. Life is still statistically much worse for non-biological children. The blood still somehow knows.
Yeah unless this actually prevents them from becoming carriers it only passes down magnified suffering to the future. Eventually humans are going to lose the ability to do this (unless you’re one of those dummies who thinks that history is eternally linear and not cyclical) and if you have propagated the gene all over the species by then, it’s going to be a serious problem.
Nature, in her typically cruel way, already provided the solution: try again.
I guess https://m.youtube.com/watch?v=k99bMtg4zRk will need a sequel soon.
Spare no expense! What could go wrong?
In the future, the phrase, “going on a blind date” may include not knowing the results of each other’s germ cell DNA compatibility test.