The gene-therapy-based treatment called Casgevy was recently approved in the UK, making it the first time that a treatment based on the CRISPR-Cas9 gene editing tool has been authorized for medical treatments. During the clinical trials, a number of patients were enrolled with either sickle cell disease (SCD) or β thalassemia, both of which are blood disorders that affect the production of healthy red blood cells. Of the 45 who enrolled for the SCD trial, 29 were evaluated in the initial 12-month efficacy assessment, with 28 of those found to be still free of the severe pain crises that characterizes SCD. For the β thalassemia trial, 42 patients were evaluated and 39 were still free of the need for red blood cell transfusions and iron chelation after the 12-month period, with the remaining three showing a marked reduction in the need for these.
Both of these blood disorders are inherited via recessive genes, meaning that in the case of SCD two abnormal copies of the β-globin (HBB) gene are required to trigger the disorder. For β thalassemia a person can be a carrier or have a variety of symptoms based on the nature of the two sets of mutated genes that involve the production of HbA (adult hemoglobin), with the severest form (β thalassemia major) requiring the patient to undergo regular transfusions. Both types of conditions have severe repercussions on overall health and longevity, with few individuals living to the age of 60.
The way that the Casgevy treatment works involves taking stem cells out of the bone marrow of the patient, after which the CRISPR-Cas9 tool is used to target the BCL11A gene and cut it out completely. This particular gene is instrumental in the switch from fetal γ globin (HBG1, HBG2) to adult β globin form. Effectively this modification causes the resulting cells to produce fetal-type hemoglobin (HbF) instead of adult HbA which would have the mutations involved in the blood disorder.
For the final step in the treatment, the modified stem cells have to be inserted back into the patient’s bone marrow, which requires another treatment to make the bone marrow susceptible to hosting the new cells. After this the patient will ideally be cured, as the stem cells produce new, HbF-producing cells that go on to create healthy hemoglobin. Although safety and costs (~US$2M per patient) considerations of such a CRISPR-Cas9 gene therapy may give pause, this has to be put against the prospect of 40-60 years of intensive symptom management.
Currently, the US FDA as well as the EU’s EMA are also looking at possibly approving the treatment, which might open the gates for similar gene-therapies.
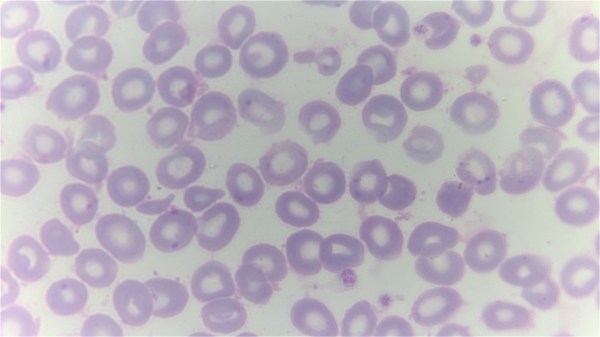
Top image: A giemsa stained blood smear from a person with beta thalassemia. Note the lack of coloring. (Credit: Dr Graham Beards, Wikimedia Commons)